Electronic structure
Overview
Teaching: 20 min
Exercises: 20 minQuestions
How can I use DFT to produce an electronic bandstructure?
How can I use DFT to produce an electronic Density of States?
Objectives
Code connection
In this episode we will use Quantum Espresso to calculate the electronic structure (bandstructure and density of states) of silicon. We will analyse and plot the electronic structure using
ase.dftandase.spectrum.
We now have all the tools in place to calculate electronic structure properties
- In the previous tutorials we created
Atomsobjects and used the Quantum Espresso calculator to get basic properties (total energies and forces) calculated using DFT. - We will now use this calculator to calculate and plot the band dispersion and density of states for silicon.
- This process takes four calculation steps; i) structure optimisation; ii) self-consistent calculation; iii) non-self-consistent calculation with sampling along a k-point path in reciprocal space; iv) non-self-consistent calculation with dense sampling in reciprocal space.
Note
This is not a course in DFT; for more details on the terminology used please see other sources.
The tocell() method can be used to build a FCC lattice
- The first step, as always, is to import libraries and build an
Atomsobject - Here we create an
FCCclass instance and then convert this to aCellobject - We will assume that the cell parameters have already been optimized.
from ase import Atoms
from ase.lattice import FCC
from ase.units import Bohr
si_fcc = Atoms(symbols='Si2',
cell = FCC(a=10.20*Bohr).tocell(),
scaled_positions=[(0.0, 0.0, 0.0),
(0.25, 0.25, 0.25)],
pbc=True)
Pseudopotential data are loaded from a local filepath
- Each pseudopotential-based electronic structure code is associated with a dataset describing the potentials used.
- In this case we specify the filepath to the SSSP pseudopotential library, and read this to get the kinetic energy cutoff energy values for silicon.
- We also store the pseudopotential for each species in the system of interest within a Python dictionary
Note
This is not a course in DFT; be aware that calculation parameters must be chosen with care for a given research problem.
from pathlib import Path
import json
pseudo_dir = Path.home() / 'SSSP_1.2.1_PBE_efficiency'
with open(str(pseudo_dir / 'SSSP_1.2.1_PBE_efficiency.json')) as fj:
dj = json.load(fj)
ecutwfc = dj['Si']['cutoff_wfc']
ecutrho = dj['Si']['cutoff_rho']
atomic_species = {'Si':dj['Si']['filename']}
Python dictionaries are used to store related calculation parameters
- There are many parameters used in a typical electronic structure calculation.
- Keywords for the
Espressoclass are specified as dictionaries grouping related parameters.controlspecifies that we want to do a self-consistent calculation, a prefix to identify the calculation, the directory where the program saves the data files, and the pseudopotentials’ location;systemspecifies the two cutoff for the FFT gridelectronspecifies the iterative diagonalization algorithm and the convergence threshold.
control = {'calculation': "scf",
'prefix': "si_fcc",
'outdir': str(Path('./si_fcc/out').absolute()) ,
'pseudo_dir': str(pseudo_dir),
'tprnfor': True,
'tstress': True}
system = {'ecutwfc': ecutwfc,
'ecutrho': ecutrho}
electrons = {'diagonalization':'davidson',
'conv_thr': 1.e-8}
EspressoProfile objects are used to hold run information
- Information for how to run QE on a particular system is stored in an
EspressoProfileobject. - Here we specify two profiles
- one where we run with 4 MPI ranks with the default parallelization
- one where we run with 4 MPI ranks using pools parallelization
from ase.calculators.espresso import EspressoProfile
plain_argv = ['mpirun', '-np', '4', 'pw.x']
pools_argv = ['-nk', '4']
profile = EspressoProfile(plain_argv)
profile_4pools = EspressoProfile(plain_argv + pools_argv)
Specify a directory for each calculation to avoid overwriting
- We are now ready to create an
Espressocalculator for the scf calculation. - As we are going to run few calculations we will specify a specific directory for each of them so that we don’t overwrite the input and standard-output files of the
pw.xprogramme. - Binary files that are read by successive calculations will be saved in the
outdirinput variable defined above. - We also specify some other key information; the Monkhort-Pack mesh for kpoints, and the offset of the MP mesh.
from ase.calculators.espresso import Espresso
scf_directory = Path('./si_fcc/scf/').absolute()
scf_calc = Espresso(directory=scf_directory,
profile=profile,
input_data={'control': control,
'system': system,
'electrons': electrons},
pseudopotentials={'Si':dj['Si']['filename']},
kpts=[8,8,8],
koffset=[1,1,1])
- We now use the calculator to run a scf calculation for our system.
si_fcc.calc = scf_calc
props_scf = si_fcc.get_properties(['energy'])
round(props_scf['energy'],8)
-215.69011555
Generate k-points path using the bandpath method
kpointpath = si_fcc.cell.bandpath(path='LGXWKG', density=20)
kpointpath.special_points
{'G': array([0., 0., 0.]),
'K': array([0.375, 0.375, 0.75 ]),
'L': array([0.5, 0.5, 0.5]),
'U': array([0.625, 0.25 , 0.625]),
'W': array([0.5 , 0.25, 0.75]),
'X': array([0.5, 0. , 0.5])}
Create a new Calculator object and Atoms for the next calculation
- First we use the dictionary
updatemethod to adjust some calculation parameters.
control.update({'calculation':"bands"})
system.update({'nbnd':12})
electrons.update({'conv_thr': 1.e-6})
- Then we setup the new calculator for bands.
- We specify a new calculation directory.
- We use the
profile_4poolsprofile, which passes the-nk 4option topw.xand distributes the band structure calculations over 4 autonomous pools.
bands_directory = Path('./si_fcc/bands').absolute()
bands_calc = Espresso(directory=bands_directory,
input_data={'control': control,
'system': system,
'electrons':electrons},
pseudopotentials=atomic_species,
profile=profile_4pools,
kpts=kpointpath)
- We copy the
Atomsobject to avoid overwriting results from the SCF calculation, attach theCalculatorand run.
si_fcc_bands = si_fcc.copy()
si_fcc_bands.calc = bands_calc
results_bands = si_fcc_bands.get_properties(['eigenvalues'])
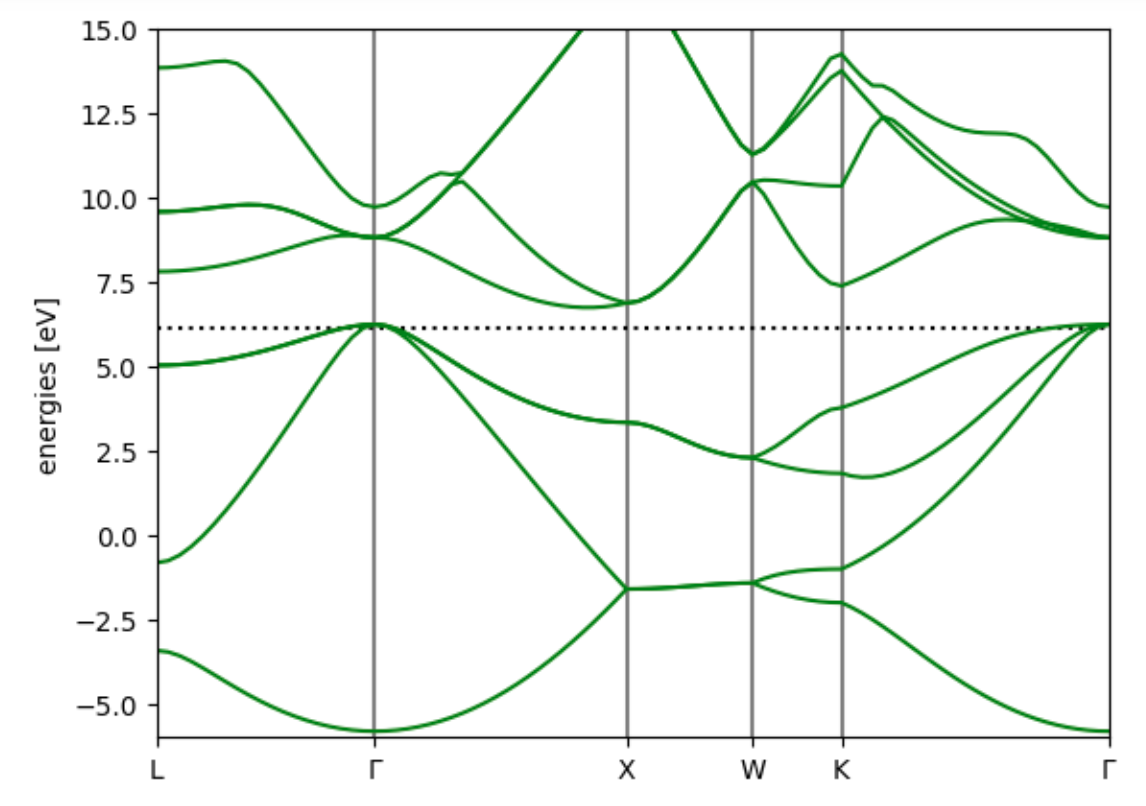
Use ase.spectrum to analyse and plot band structures
- Create a
BandStructureobject holding the k-point path and eigenvalues. - Use as reference level the Fermi level that has been computed in the scf calculation.
from ase.spectrum.band_structure import BandStructure
si_fcc_band_structure = BandStructure(path=kpointpath,
energies=results_bands['eigenvalues'], reference=si_fcc.calc.get_fermi_level())
- The
Bandstructureobject has methods for plotting and saving.
bandplot = si_fcc_band_structure.plot(emin=-6, emax=15)
bandplot.figure.savefig('band_structure_Si.png')
Use ase.dft to compute the Density of States
- As for the bandstructure calculation, we create a new
Calculator,Atomsand calculation directory for this calculation. - We change verbosity to ‘high’ because printout of energies in the standard output wound be suppressed for the large number of kpoints we are using.
control.update({'calculation':'nscf',
'verbosity': 'high'})
system.update({'nbnd': 20})
nscf_directory = Path('./si_fcc/nscf').absolute()
nscf_calc = Espresso(directory =nscf_directory,
profile = profile_4pools,
input_data={'control': control,
'system': system,
'electron': electrons},
pseudopotentials=atomic_species,
kpts=[16,16,16],
koffset=[0,0,0])
si_fcc_nscf = si_fcc.copy()
si_fcc_nscf.calc = nscf_calc
nscf_results = si_fcc_nscf.get_properties(['eigenvalues'])
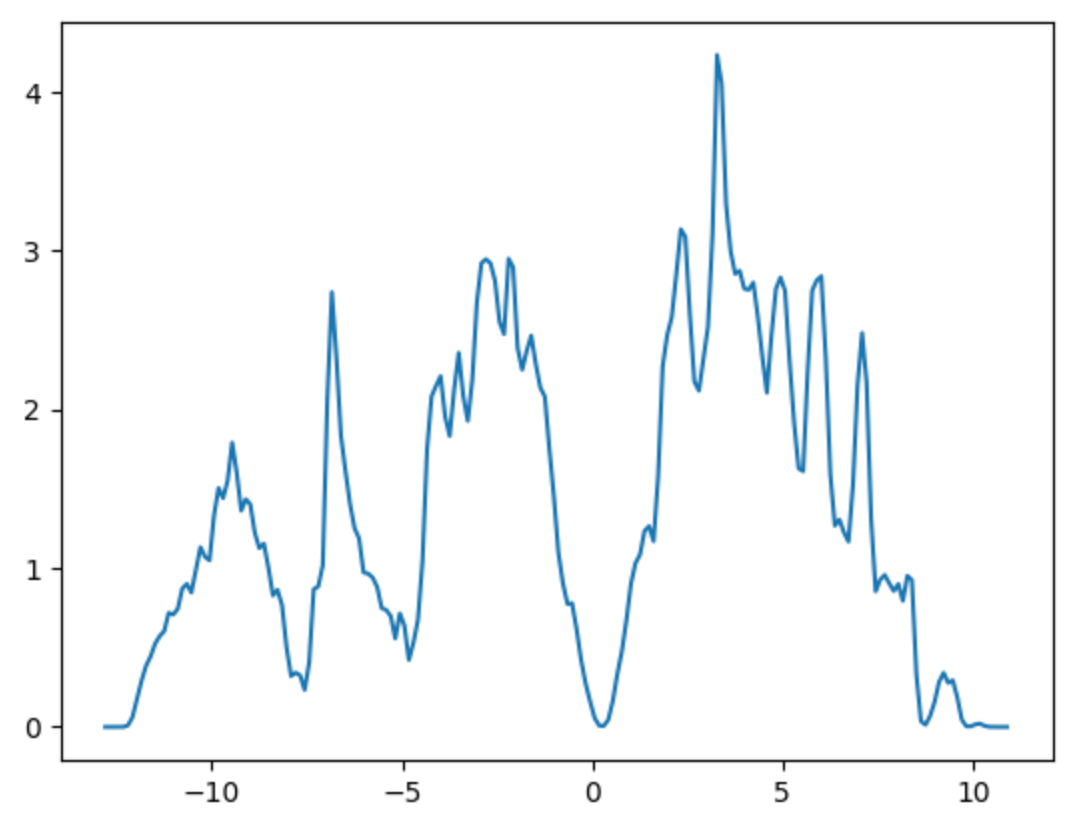
- We use ASE’s
DOSclass to compute the Density of States.
from ase.dft.dos import DOS
from matplotlib import pyplot as plt
dos = DOS(nscf_calc, npts = 200,width=0.2)
- This is initialised using the
Calculationobjectnscf_calc. - We can then access the
energiesproperty andget_dos“getter” method needed for plotting the DOS.
energies=dos.energies
dosvals = dos.get_dos()
plt.plot(energies, dosvals)
plt.axis([-13, 15, 0, 5])
plt.savefig('DOS_Si.png')
Key Points
We now have all the tools in place to calculate electronic structure properties
Use the
tocell()method to build a FCC latticePseudopotential data are loaded from a local filepath
Python dictionaries are used to store related calculation parameters
EspressoProfileobjects are used to hold run informationSpecify a directory for each calculation to avoid overwriting
Generate k-points path using the
bandpathmethodCreate a new
Calculatorobject andAtomsfor the next calculationUse
ase.spectrumto analyse and plot band structuresUse
ase.dftto compute the Density of States