External calculators
Overview
Teaching: 15 min
Exercises: 20 minQuestions
How can I calculate standard properties using an external calculator?
How can I calculate standard properties using a machine learned potential?
What is a convenient way to test for k-point convergence?
Objectives
Calculate properties using an external calculator and machine learned potential
Perform a convergence test for density-functional theory calculation parameters
Code connection
In this episode we explore two external calculators: Quippy, which provides an interface to a range of interatomic and tight-binding potentials, including Gaussian Approximation Potentials, and the DFT calculator GPAW.
The quippy package provides a Python interface to a range of interatomic and tight-binding potentials
- Some calculators have interfaces which are not packaged with ASE, but available elsewhere.
- For example, the
quippypackage provides a Python interface to a range of interatomic and tight-binding potentials. - In this episode we apply a machine-learning-based potential for Si.
Getting the model and training data
The documentation for Gaussian Approximation Potentials (GAP) links to a few published GAP models. To download and extract the data in a Jupyter notebook you can use bash.
%%bash wget -q https://www.repository.cam.ac.uk/bitstream/handle/1810/317974/Si_PRX_GAP.zip unzip Si_PRX_GAP.zip -d ./Si_PRX_GAP
Note
This requires a version of quippy that includes GAP. At the moment,
pip installseems to work better than installing from conda-forge.
The workflow for external, file-based and built-in calculators is the same
- First, we import libraries and create an
Atomsobject; in this case, a silicon supercell.
from quippy.potential import Potential
si = ase.build.bulk('Si') * 4
- Second we attach a calculator, in this case a
Potentialobject imported fromquippy.
si.calc = Potential(param_filename='./Si_PRX_GAP/gp_iter6_sparse9k.xml')
- Third, we calculate an energy; in this case we place this in a
forloop and apply a random walk to the positions.
import matplotlib.pyplot as plt
energies = []
for _ in range(10):
si.rattle(stdev=0.01)
energies.append(si.get_potential_energy())
fig, ax = plt.subplots()
ax.plot(energies, 'o-')
ax.set_xlabel('Random walk step')
ax.set_ylabel('Energy / eV')
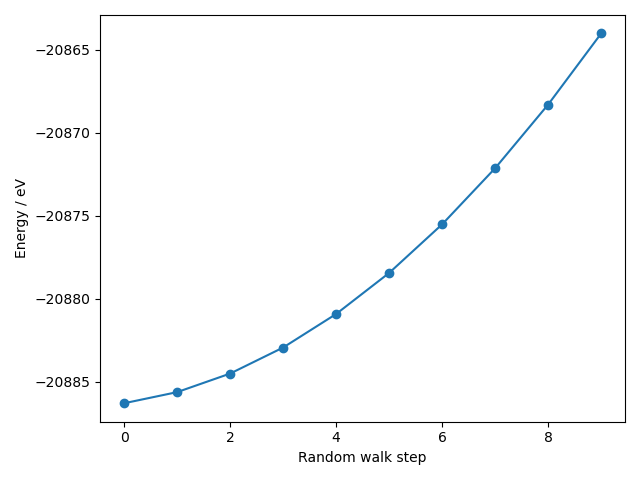
- This is a bit more expensive than EMT but still a lot cheaper than density-functional theory!
- A lot of work goes into developing a new potential, but with tools like quippy and ASE it is fairly easy for researchers to pick up the resulting model and apply it.
GPAW is a package for electronic structure calculations which relies on ASE
- GPAW is an electronic structure code implemented as a Python library with C backend.
- GPAW includes a few command line tools, but is generally always used with ASE.
- Due to this close relationship many GPAW developers are also ASE developers.
- GPAW implements the projector-augmented wave (PAW) method which requires atomic “setups” with pseudopotentials.
Getting the data
To download the necessary pseudopotential data, run
gpaw install-data ./gpaw-pot/in the terminal and typeywhen prompted`.
You can use GPAW to perform a DFT calculation from the Python shell
- To begin with, we run a single-point energy calculation using Kohn-Sham density-functional theory (DFT).
- First, we import GPAW and create the atoms object
from gpaw import GPAW, PW
atoms = ase.build.bulk('Cu')
- Second, we attach GPAW as a calculator
- We specify a number of parameters:
xcsets the exchange-correlation functionalkptssets the Brillouin-zone samplingmodesets the basis set; in this case a 400 eV cutoff plane-wave basis is used.
- For more information about valid parameters, see the GPAW docs.
atoms.calc = GPAW(xc='PBE', kpts=(3, 3, 3), mode=PW(400)) - Finally, we run the calculation and get the result
energy = atoms.get_potential_energy()
___ ___ ___ _ _ _
| | |_ | | | |
| | | | | . | | | |
|__ | _|___|_____| 22.8.0
|___|_|
User: adam@Arctopus
Date: Tue Apr 4 11:30:25 2023
Arch: x86_64
Pid: 97252
CWD: /home/adam/src/ase-tutorial-2023/development
Python: 3.10.0
gpaw: /home/adam/.conda/envs/user-base/envs/ase-tutorials/lib/python3.10/site-packages/gpaw
_gpaw: /home/adam/.conda/envs/user-base/envs/ase-tutorials/lib/python3.10/site-packages/
_gpaw.cpython-310-x86_64-linux-gnu.so
ase: /home/adam/src/ase/ase (version 3.23.0b1-70eab133b6)
numpy: /home/adam/.conda/envs/user-base/envs/ase-tutorials/lib/python3.10/site-packages/numpy (version 1.23.5)
scipy: /home/adam/.conda/envs/user-base/envs/ase-tutorials/lib/python3.10/site-packages/scipy (version 1.10.1)
libxc: 5.2.3
units: Angstrom and eV
cores: 1
OpenMP: True
OMP_NUM_THREADS: 1
Input parameters:
kpts: [3 3 3]
mode: {ecut: 400.0,
name: pw}
xc: PBE
System changes: positions, numbers, cell, pbc, initial_charges, initial_magmoms
Initialize ...
It is important to check for convergence of the energy with respect to k-point sampling
- Using a
forloop we can re-run the calculation for a increasing k-point mesh densities.- In this case we pass a dictionary specification that generates a mesh that is shifted off the Gamma-point.
- We also use the
txtkeyword to direct output to a text file.
- We collect timing information to understand how increasing mesh density impacts calculation cost.
- The argument
str(k)is the timer name
- The argument
- The number of k-points is retrieved with
get_ibz_k_points(). Note thatget_bz_k_points()is not the correct function to use as this does not include reductions made due to system symmetry.
Note
This will take a few minutes to run: to see live output, open a terminal and use
tail -f kpts_serial.txtto see this file grow. When it is finished, you can exittailwith ctrl-c.
from ase.utils.timing import Timer
timer = Timer()
energies, times, nkpts = [], [], []
for k in range(3,9):
atoms.calc = GPAW(mode=PW(400), xc='PBE',
kpts={'size': [k, k, k],
'gamma': False},
txt='kpts_serial.txt')
timer.start(str(k))
energies.append(atoms.get_potential_energy())
timer.stop(str(k))
times.append(timer.get_time(str(k)))
nkpts.append(len(atoms.calc.get_ibz_k_points()))
Timing: incl. excl.
-----------------------------------------------------------
Hamiltonian: 0.096 0.000 0.0% |
Atomic: 0.088 0.001 0.0% |
XC Correction: 0.087 0.087 0.8% |
Calculate atomic Hamiltonians: 0.001 0.001 0.0% |
Communicate: 0.000 0.000 0.0% |
Initialize Hamiltonian: 0.000 0.000 0.0% |
Poisson: 0.000 0.000 0.0% |
XC 3D grid: 0.007 0.007 0.1% |
LCAO initialization: 0.344 0.082 0.7% |
LCAO eigensolver: 0.147 0.000 0.0% |
Calculate projections: 0.000 0.000 0.0% |
DenseAtomicCorrection: 0.000 0.000 0.0% |
Distribute overlap matrix: 0.000 0.000 0.0% |
Orbital Layouts: 0.001 0.001 0.0% |
Potential matrix: 0.144 0.144 1.3% ||
Sum over cells: 0.001 0.001 0.0% |
LCAO to grid: 0.018 0.018 0.2% |
Set positions (LCAO WFS): 0.097 0.012 0.1% |
Basic WFS set positions: 0.002 0.002 0.0% |
Basis functions set positions: 0.000 0.000 0.0% |
P tci: 0.014 0.014 0.1% |
ST tci: 0.039 0.039 0.3% |
mktci: 0.030 0.030 0.3% |
PWDescriptor: 0.017 0.017 0.2% |
SCF-cycle: 1.907 0.101 0.9% |
Davidson: 0.459 0.152 1.3% ||
Apply H: 0.033 0.027 0.2% |
HMM T: 0.006 0.006 0.1% |
Subspace diag: 0.076 0.004 0.0% |
calc_h_matrix: 0.056 0.023 0.2% |
Apply H: 0.034 0.027 0.2% |
HMM T: 0.006 0.006 0.1% |
diagonalize: 0.009 0.009 0.1% |
rotate_psi: 0.006 0.006 0.1% |
calc. matrices: 0.160 0.091 0.8% |
Apply H: 0.069 0.056 0.5% |
HMM T: 0.012 0.012 0.1% |
diagonalize: 0.023 0.023 0.2% |
rotate_psi: 0.014 0.014 0.1% |
Density: 0.191 0.000 0.0% |
Atomic density matrices: 0.019 0.019 0.2% |
Mix: 0.064 0.064 0.6% |
Multipole moments: 0.001 0.001 0.0% |
Pseudo density: 0.106 0.015 0.1% |
Symmetrize density: 0.091 0.091 0.8% |
Hamiltonian: 1.152 0.004 0.0% |
Atomic: 0.952 0.015 0.1% |
XC Correction: 0.937 0.937 8.2% |--|
Calculate atomic Hamiltonians: 0.006 0.006 0.1% |
Communicate: 0.000 0.000 0.0% |
Poisson: 0.002 0.002 0.0% |
XC 3D grid: 0.189 0.189 1.7% ||
Orthonormalize: 0.003 0.000 0.0% |
calc_s_matrix: 0.001 0.001 0.0% |
inverse-cholesky: 0.000 0.000 0.0% |
projections: 0.001 0.001 0.0% |
rotate_psi_s: 0.000 0.000 0.0% |
Set symmetry: 0.025 0.025 0.2% |
Other: 8.970 8.970 79.0% |-------------------------------|
-----------------------------------------------------------
Total: 11.359 100.0%
Memory usage: 390.56 MiB
Date: Tue Apr 4 11:30:36 2023
- Finally, we plot the results using a matplotlib figure with multiple subplots.
fig, axes = plt.subplots(nrows=2, sharex=True)
axes[0].plot(nkpts, energies, 'o-')
axes[0].set_ylabel('energy / eV')
axes[1].plot(nkpts, times, 'o-')
axes[1].set_ylabel('Calculation time / s')
axes[1].set_ylim([0, None])
axes[1].set_xlabel('number of k-points')
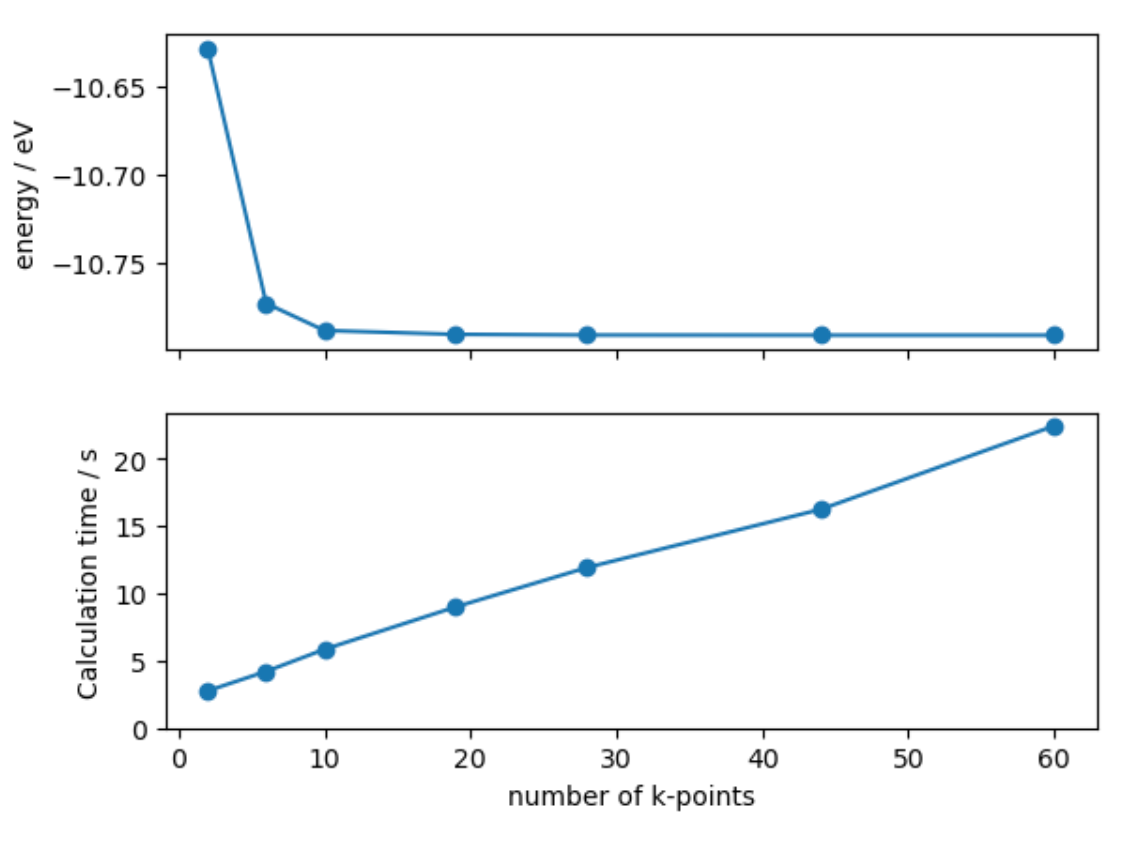
- We find that the computational cost per k-point is roughly linear, but the energy convergence is relatively slow.
import numpy as np
change_in_energies = np.diff(energies)
print(change_in_energies)
[ 0.04294983 -0.05835439 0.00339695 -0.00389174 -0.00460936]
Key Points
The
quippypackage provides a Python interface to a range of interatomic and tight-binding potentialsThe workflow for external, file-based and built-in calculators is the same
GPAW is a package for electronic structure calculations which relies on ASE
You can use GPAW to perform a DFT calculation from the Python shell
It is important to check for convergence of DFT energies with respect to k-point sampling