Built-in calculators
Overview
Teaching: 20 min
Exercises: 25 minQuestions
How can I calculate standard properties using a built-in calculator?
How can I fit simple models to my calculations?
What is the best way to store and share property data?
Objectives
Calculate properties using a built-in
CalculatorobjectIdentify the optimum bonding length for a simple metallic system
Code connection
In this episode we explore the
ase.calculatorse.emtmodule andase.calculatorse.ljmodule, both of which provide built-in tools for calculating standard properties (energy, forces and stress) from a set of atomic positions.
Atoms objects can calculate properties using an attached “Calculator”
- In ASE,
Atomsobjects can try to calculate or fetch properties using an attachedCalculator. - In this tutorial we will make tour of a variety of Calculators, highlighting some of the differences between them.
- A master list of the available Calculators can be found here.
Properties of metal alloy systems can be calculated using Effective Medium Theory
- Much of the ASE documentation and tutorials makes use of the built-in “EMT” calculator
- This is because it is convenient and “fast enough”
- EMT implements the Effective Medium Theory potential for Ni, Cu, Pd, Ag, Pt and Au.
- Some other elements are included “for fun”, but really this is a method for alloys of those metals.
Warning
If you want to do a real application using EMT, you should use the much more efficient implementation in the ASAP calculator.
Calculators can calculate properties in three easy steps
- EMT can be used to calculate, for example, the energy of an infinite gold wire.
- The first step is to create an
Atomsobject describing a gold wire which is infinite in the x-direction.
Python tip
You may not recognise or understand the syntax used in the function definition, for example
spacing: float = 2.5. These are optional Type Hints, which were added in Python 3.5 (2015) and are becoming more widely-used as support is dropped for older versions.
from ase import Atoms
from ase.calculators.emt import EMT
from ase.visualize import view
def make_wire(spacing: float = 2.5,
box_size: float = 10.0) -> Atoms:
wire = Atoms('Au',
positions=[[0., box_size / 2, box_size / 2]],
cell=[spacing, box_size, box_size],
pbc=[True, False, False])
return wire
atoms = make_wire()
view(atoms, viewer='ngl')
- The second step is to attach the calculator of choice, in this case
EMT()
atoms.calc = EMT()
- The third and final step is to call a “getter” method for the property of choice, in this case the potential energy.
energy = atoms.get_potential_energy()
print(f"Energy: {energy} eV")
Energy: 0.9910548478768826 eV
Discussion
Why did we need the parentheses () in the line
atoms.calc = EMT()?
Scientific Python libraries allow us to fit models to our calculations
- We can use the workflow above to investigate how energy varies with the atom spacing and fit a model.
- First we wrap the workflow up within a single function which returns the energy for a given atom spacing.
Python tip
if you need to apply a function to each element of some data,
mapcan provide an elegant alternative to for-loops and list comprehensions!
import numpy as np
distances = np.linspace(2., 3., 21)
def get_energy(spacing: float) -> float:
atoms = make_wire(spacing=spacing)
atoms.calc = EMT()
return atoms.get_potential_energy()
energies = list(map(get_energy, distances))
- Second we use Numpy to fit a polynomial to the generated data
from numpy.polynomial import Polynomial
fit = Polynomial.fit(distances, energies, 3)
- Finally we use Matplotlib to visualise the data and fit
IPython Magics
%matplotlib inlineturns on “inline plotting”, where plots will appear in your notebook rather than as a separate pop-out window. This is our first example of an IPython magic command. You will see more examples later in the tutorial.
%matplotlib inline
import matplotlib.pyplot as plt
fig, ax = plt.subplots()
x = np.linspace(2., 3., 500)
_ = ax.plot(distances, energies, 'o', label='calculated')
_ = ax.plot(x, fit(x), '-', label='cubic')
_ = ax.legend()
_ = ax.set_xlabel('Spacing / Å')
_ = ax.set_ylabel('Energy / eV')
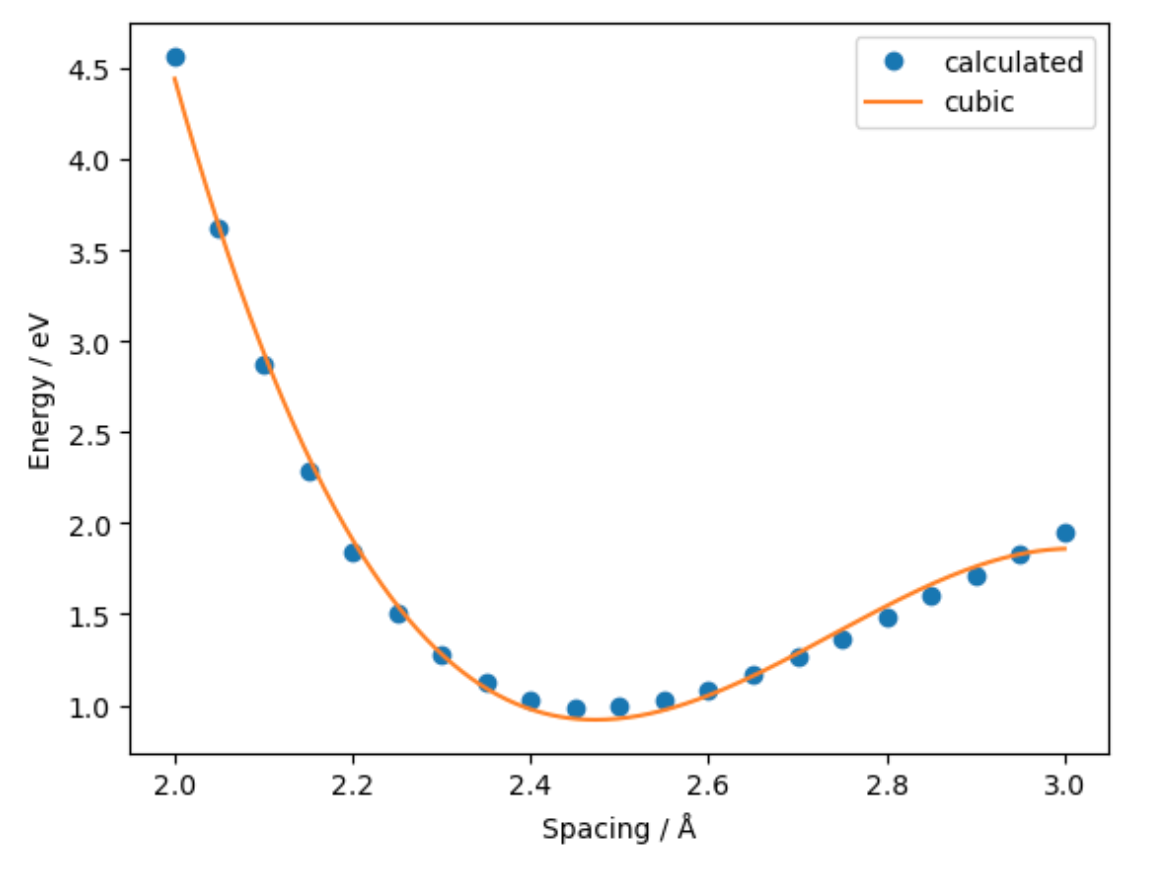
Exercise: Equation of State for bulk gold
The plot above resembles the Equation-of-State (EOS) curve for a solid. Using a similar workflow and the
EquationOfStateclass, fit an equation of state to bulk gold and obtain an equilibrium volume.
The EMT Calculator can also be used to obtain forces and unit cell stress
print("Forces: ")
print(atoms.get_forces())
print("Stress: ")
print(atoms.get_stress())
Forces:
[[0. 0. 0.]]
Stress:
[ 0.00396458 -0. -0. -0. -0. -0. ]
Discussion
Why are the forces exactly zero for this system?
- We can check which properties are implemented by a particular calculator by inspecting the
implemented propertiesattribute:
print(EMT.implemented_properties)
['energy', 'free_energy', 'energies', 'forces', 'stress', 'magmom', 'magmoms']
Discussion
Why do we not need to include parenthesis () here? Do we expect
EMT().implemented_propertiesto work as well asEMT.implemented_properties?
Where possible request a standalone set of property data
- It is sometimes convenient to have properties attached to a particular
Calculatorobject. - For example, forces are used heavily by dynamics and optimizer routines, as we will see in the next tutorial.
- However for Open Science purposes it is easier to store and share data that is not connected to a
Calculator. This is because Calculators might depend on a particular machine environment, memory state or software license. - We can request a standalone set of property data with
get_properties.
properties = atoms.get_properties(['energy', 'forces', 'stress'])
print(properties)
(Properties({'energy': 0.9910548478768826, 'natoms': 1, 'energies': array([0.99105485]), 'free_energy': 0.9910548478768826, 'forces': array([[0., 0., 0.]]), 'stress': array([ 0.00396458, -0. , -0. , -0. , -0. ,
-0. ])})
- Importantly, this will not change even if the
Atomsobject is modified and properties are recalculated.
Warning
This is a new feature and does not yet work well for all calculators.
The Lennard-Jones potential can be used to model the interaction between two non-bonding atoms or molecules
- The classic Lennard-Jones potential is implemented in
ase.calculators.lj. - You can set the ε and σ parameters in the Calculator constructor:
from ase.calculators.lj import LennardJones
l = 4.1
atoms = Atoms('Xe2',
positions=[[0., 0., -l / 2],
[0., 0., l / 2]],
pbc=False)
atoms.calc = LennardJones(sigma=(4.1 / 2**(1/6)))
atoms.get_forces()
array([[ 0.00000000e+00, 0.00000000e+00, -6.49886649e-16],
[ 0.00000000e+00, 0.00000000e+00, 6.49886649e-16]])
Discussion
Why are the forces so low at this geometry?
Exercise: Lennard-Jones binding curve
Try varying the distance between the atoms. Can you reproduce the classic plot of a Lennard-Jones binding curve?
Key Points
Atoms objects can calculate properties using an attached
CalculatorProperties of metal alloy systems can be calculated using Effective Medium Theory
Calculators can calculate properties in three easy steps
Scientific Python libraries allow us to fit models to our calculations
The EMT Calculator can also be used to obtain forces and unit cell stress
Where possible request a standalone set of property data
The Lennard-Jones potential can be used to model the interaction between two non-bonding atoms or molecules