Building and Manipulating Atoms
Overview
Teaching: 25 min
Exercises: 25 minQuestions
How can I build molecules and bulk structures more efficiently?
How can I create supercells and point defects?
Objectives
Build a molecule using the built-in database
Build a crystal using built-in crystal structure types
Build (optimal) supercell expansions
Remove, add or swap atom(s) to create point defects
Code connection
In this episode we explore the
ase.buildmodule, which contains tools for building structures using parameters rather than detailed lists of positions.
A set of simple molecules are pre-defined in ASE
- Definitions for a set of simple molecules (the “G2” set and the “S22” set) are included with ASE.
- So in fact the easiest way to get an N2 molecule is with
ase.build.molecule.
import ase.build
from ase.visualize import view
g2_n2 = ase.build.molecule('N2')
view(g2_n2, viewer='ngl')
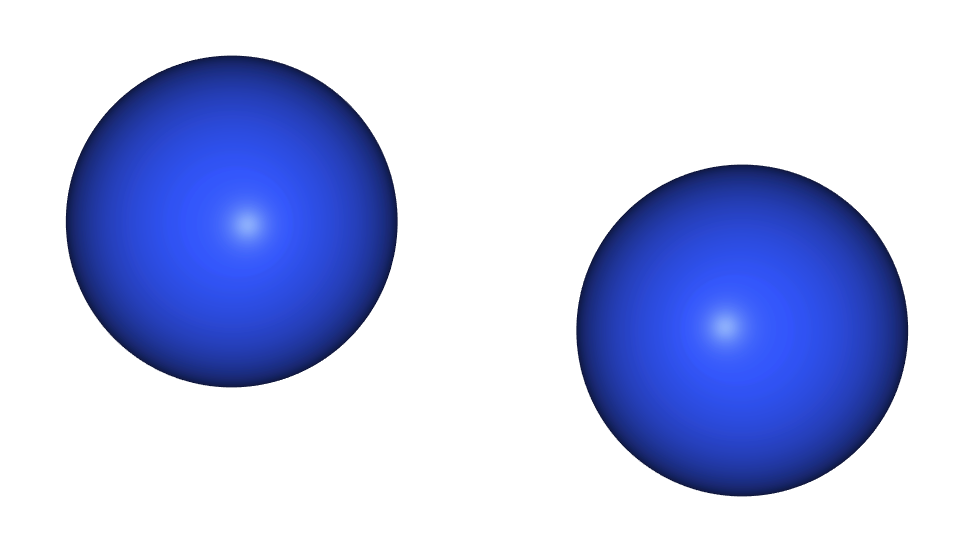
- If we inspect the type of
g2_n2we see that it is anAtomsobject, just like that we created manually in earlier episodes.
type(g2_n2)
ase.atoms.Atoms
- And it’s just as easy to get more interesting things like a buckyball!
view(ase.build.molecule('C60'), viewer='ngl')

There are pre-defined lattice parameters and crystal types to create bulk systems
- The equivalent tool to build crystals is
ase.build.bulk. - This includes lattice parameters for some elemental reference states; the list is in the code here.
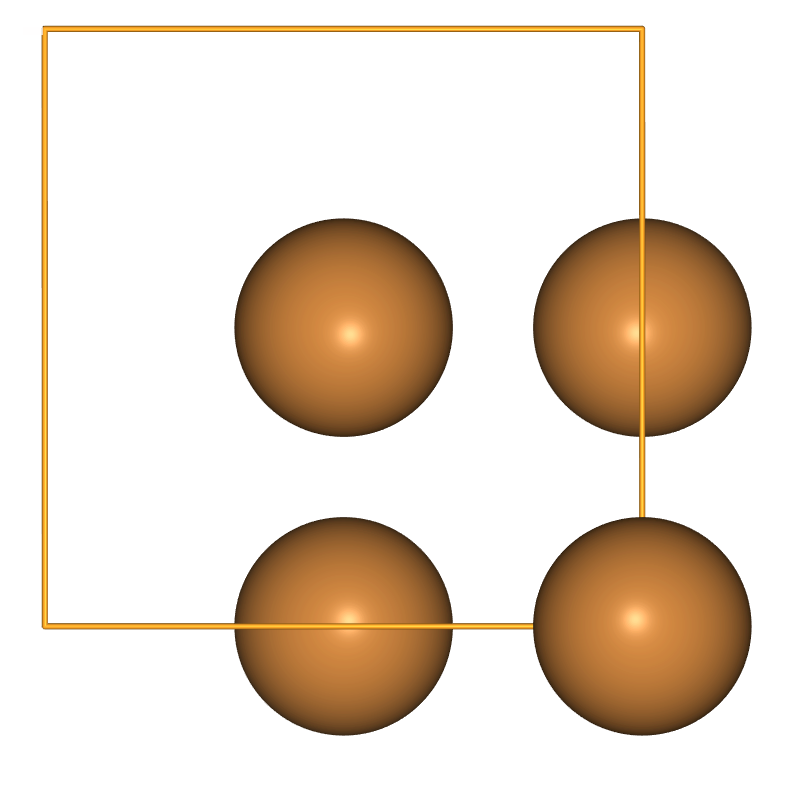
- So we get copper, for example, with very little work:
view(ase.build.bulk('Cu', cubic=True), viewer='ngl')
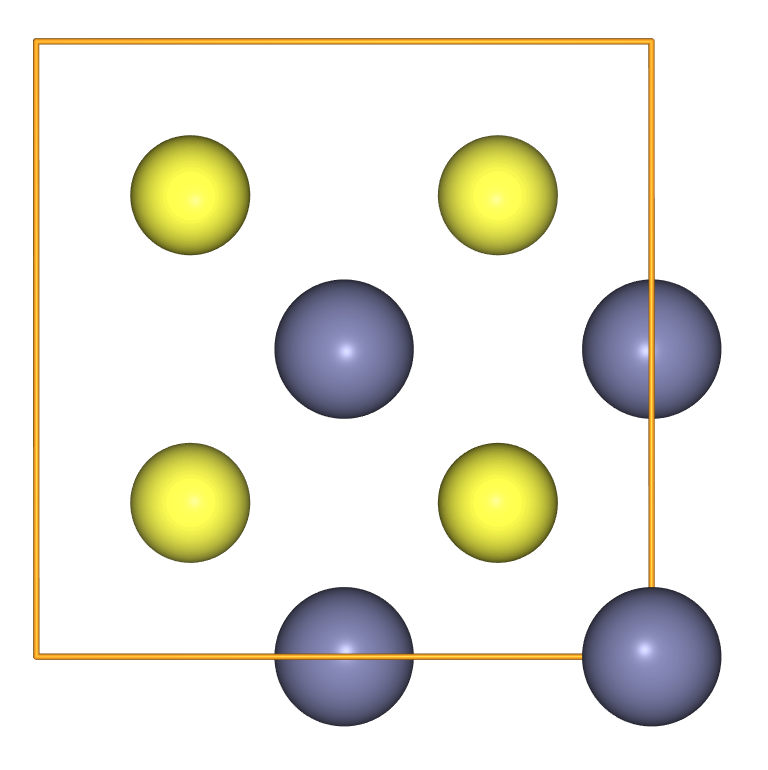
- To create ZnS in the zincblende structure we have to provide the
crystalstructureand the lattice parametera.
view(
ase.build.bulk('ZnS',
crystalstructure='zincblende',
a=5.387,
cubic=True),
viewer='ngl'
)
A compact notation can be used to create a supercell
- Supercell expansions of a unit cell are commonly used when modelling, for example, lattice dynamics or defect systems.
- A compact notation can be used to create the repeated unit cell.
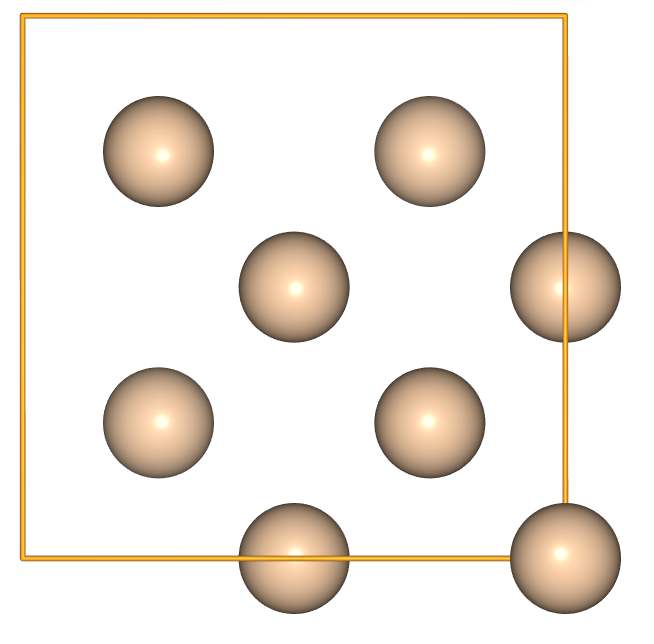
- Start by creating a cubic unit cell of Si.
si = ase.build.bulk('Si', cubic=True)
view(si, viewer='ngl')
- Use integer multiplication to perform equal repetition in each direction:
view(si * 4, viewer='ngl')

- Use a 3-list or 3-tuple to perform unequal repetitions in each direction:
view(si * [2, 4, 1], viewer='ngl')
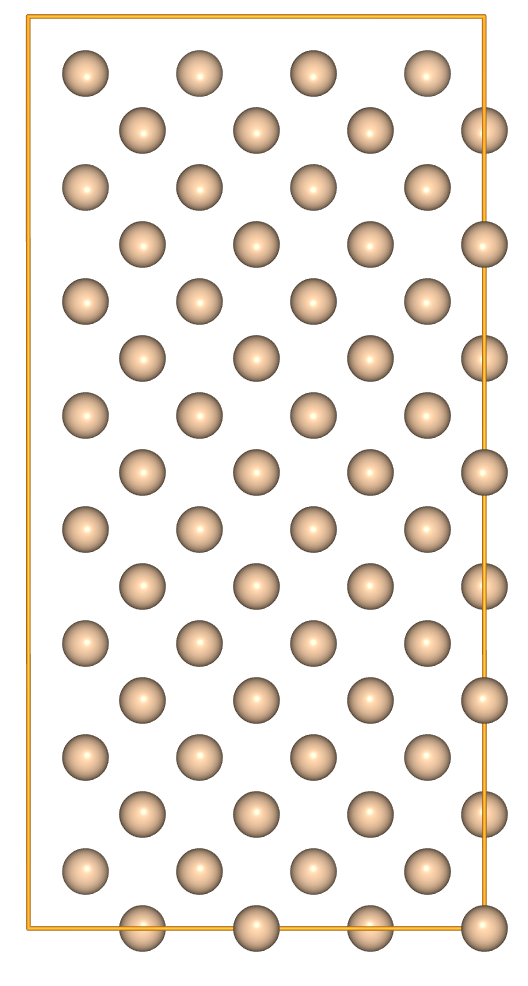
Cell parameters can be inspected using the Atoms.cell attribute
- When we model dilute defects it can be useful to use cubic supercell expansions, as these will maximise the minimum distance between periodic images.
- Our starting point for this is often a non-cubic primitive cell.
si_prime = ase.build.bulk('Si')
view(si_prime, viewer='ngl')
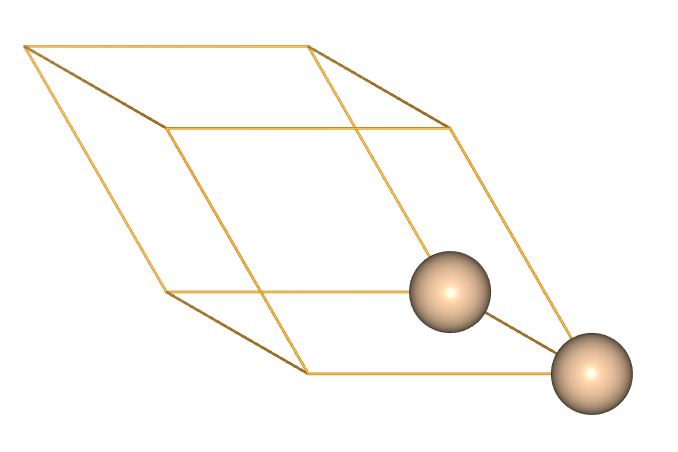
- We can inspect the cell parameters using the
Atoms.cellattribute.
si_prime.cell
Cell([[0.0, 2.715, 2.715], [2.715, 0.0, 2.715], [2.715, 2.715, 0.0]])
- We find that
Atoms.cellreturns an instance of the ASECellclass. - The
Cellobject represents three lattice vectors forming a parallel epiped.
ASE can search for the matrix which gives the most cubic supercell
- Once we have a primitive cell, we can perform a numerical search to find the optimal 3x3 array for forming the most cubic supercell; this process may take a few seconds.
- We specify three positional arguments: unit cell parameters, the target size of the supercell, and the target shape. For more information, you can inspect the docstring.
Academic background
This algorithm is described in more detail in the ASE docs, and was developed to support supercell doping calculations
optimal_array = ase.build.find_optimal_cell_shape(si_prime.cell, 4, 'sc', verbose=True)
target metric (h_target):
[[1. 0. 0.]
[0. 1. 0.]
[0. 0. 1.]]
normalization factor (Q): 0.184162
idealized transformation matrix:
[[-1. 1. 1.]
[ 1. -1. 1.]
[ 1. 1. -1.]]
closest integer transformation matrix (P_0):
[[-1 1 1]
[ 1 -1 1]
[ 1 1 -1]]
smallest score (|Q P h_p - h_target|_2): 0.000000
optimal transformation matrix (P_opt):
[[-1 1 1]
[ 1 -1 1]
[ 1 1 -1]]
supercell metric:
[[5.43 0. 0. ]
[0. 5.43 0. ]
[0. 0. 5.43]]
determinant of optimal transformation matrix: 4
- We can then pass the 3x3 array to
ase.build.make_supercellto form a cubic supercell.
cubic_Si_expansion = ase.build.make_supercell(si_prime, optimal_array)
view(cubic_Si_expansion, viewer='ngl')
cubic_Si_expansion.cell
Cell([5.43, 5.43, 5.43])
- We find that, as expected, this is equal to the Si cubic crystal structure pre-defined in ASE.
si = ase.build.bulk('Si', cubic=True)
si.cell
Cell([5.43, 5.43, 5.43])
An Atoms object can be treated as an array of Atom objects
- An
Atomsobject can be treated as a list ofAtomobjects which we can iterate over. Atomis a class for representing….you guessed it…a single atom.
crystal = ase.build.bulk('ZnS',
crystalstructure='zincblende',
a=5.387,
cubic=True)
for atom in crystal:
print(atom.symbol, atom.position, atom.mass)
Zn [0. 0. 0.] 65.38
S [1.34675 1.34675 1.34675] 32.06
Zn [0. 2.6935 2.6935] 65.38
S [1.34675 4.04025 4.04025] 32.06
Zn [2.6935 0. 2.6935] 65.38
S [4.04025 1.34675 4.04025] 32.06
Zn [2.6935 2.6935 0. ] 65.38
S [4.04025 4.04025 1.34675] 32.06
- Numpy-like array indexing and slicing is also available.
- For example, we can combine indexing with list comprehension to return the zinc sub-lattice.
zinc_indices = [i for i, atom in enumerate(crystal) if atom.symbol == 'Zn']
zinc_sublattice = crystal[zinc_indices]
view(zinc_sublattice, viewer='ngl')
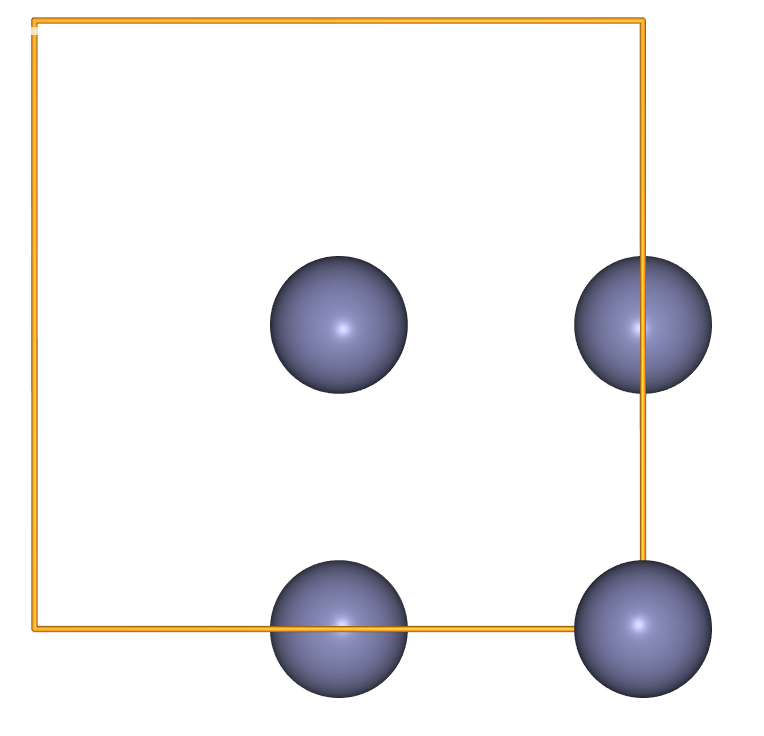
- When we index multiple atoms, an
Atomsobject is returned.
type(zinc_sublattice)
ase.atoms.Atoms
Methods and operations can be used to create point defects
- An individual
Atomcan be appended to create an interstitial defect.
from ase import Atom
composite = zinc_sublattice.copy()
composite.append(Atom('He', position=(1.34675, 4.04025, 4.04025)))
view(composite, viewer='ngl')
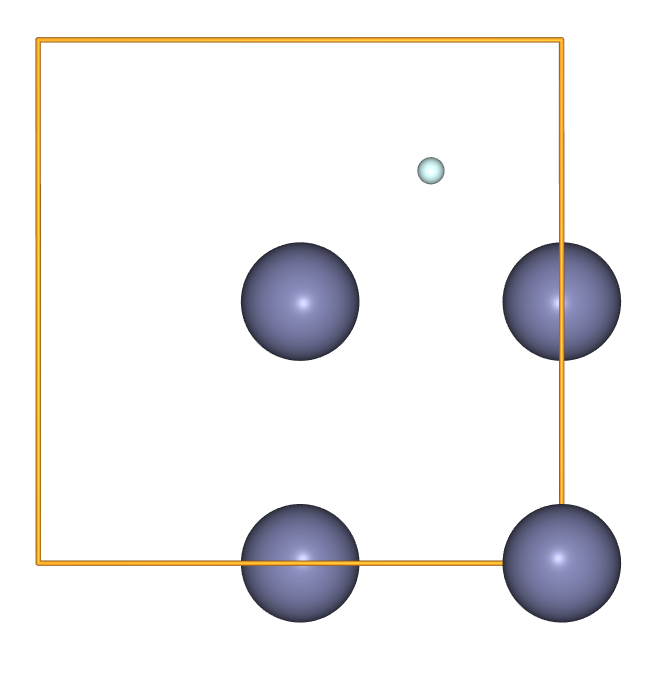
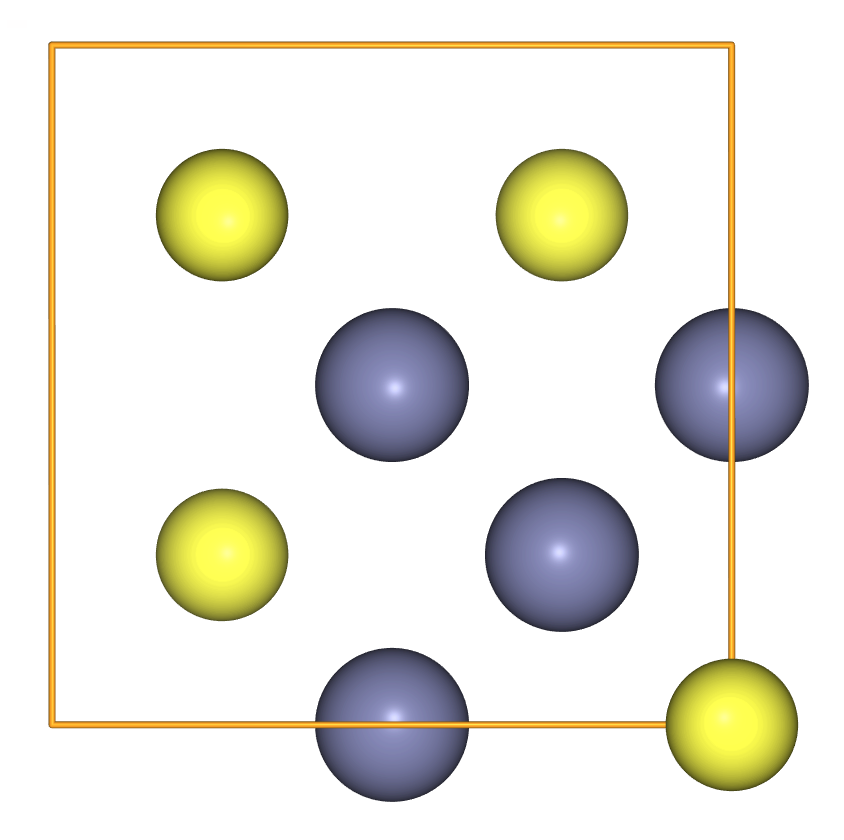
- While
Atomsis not exactly a regular Python data collection, it plays nicely with the delete operation. - We can use this operation to create, for example, a zinc-vacancy defect:
zinc_vacancy = crystal.copy()
del zinc_vacancy[0]
view(zinc_vacancy, viewer='ngl')
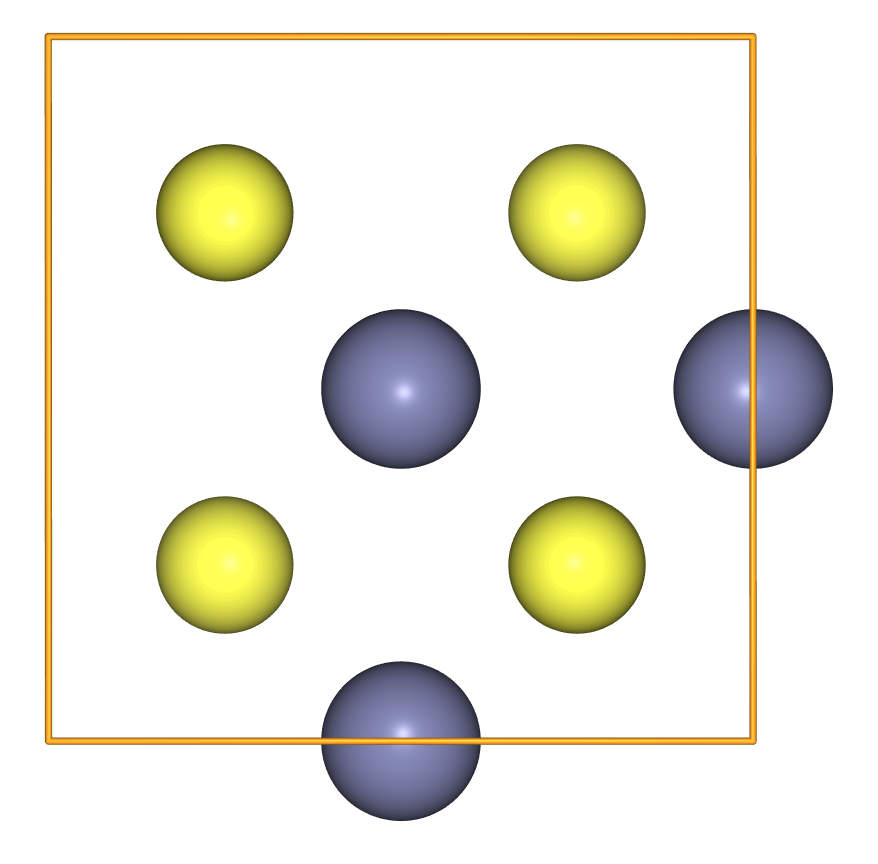
- To create antisite disorder, we can swap two positions from the positions array.
antisite = crystal.copy()
antisite.positions[[0, 1]] = antisite.positions[[1, 0]]
view(antisite, viewer='ngl')
Exercise: Distorted sphalerite
It is possible to combine entire
Atomswith+. In this case, the firstAtomstakes precedence to determine the cell parameters. Use this to create a distorted sphalerite cell, with the S sublattice translated along the x-coordinate relative to the Zn sublatticeSolution
sulfur_indices = [i for i, atom in enumerate(crystal) if atom.symbol == 'S'] sulfur_sublattice = crystal[sulfur_indices] sulfur_sublattice.translate([.3, 0., 0.]) view(zinc_sublattice + sulfur_sublattice, viewer='ngl')
Exercise: Water animation
Create an animation of a water molecule being wrapped in a C60 cage (or something even cooler!)
Hints:
- The GIF animation will need to be generated with a list of Atoms objects
- Molecules can be combined with +
- To get the wrapping effect we need to keep adding atoms that are near to the atoms already added
- To avoid writing too much repetitive code, use Python’s looping tools
Solution
water = ase.build.molecule('H2O') water.center() bucky = ase.build.molecule('C60') bucky.center() start_atom = 36 distances = bucky.get_all_distances()[start_atom] sorted_bucky_indices = sorted(enumerate(distances), key = (lambda x: x[1])) sorted_bucky_indices = [i for i, _ in sorted_bucky_indices] sorted_bucky = bucky[sorted_bucky_indices] frames = [water.copy()] for i in range(len(sorted_bucky)): frames.append(water + sorted_bucky[:i + 1]) from ase.io.animation import write_gif _ = write_gif('wrapped_molecule.gif', frames)
Key Points
A set of simple molecules are pre-defined in ASE
There are pre-defined lattice parameters and crystal types to create bulk systems
A compact notation can be used to create a supercell
Cell parameters can be inspected using the
Atoms.cellattributeASE can search for the matrix which gives the most cubic supercell
An
Atomsobject can be treated as an array ofAtomobjectsMethods and operations can be used to create point defects