Local optimisation
Overview
Teaching: 20 min
Exercises: 30 minQuestions
How can I find kinetically stable structures?
Is it possible to apply constraints to my system?
How can I optimise other degrees of freedom beyond atom positions?
Can I generate amorphous structures using ASE?
Objectives
Perform geometry optimization using MOPAC
Perform molecular dynamics and geometry optimisation using a machine learned potential
Apply constraints to an optimisation routine
Code connection
In this episode we will use the
ase.optimizemodule to perform geometry optimisation, andase.constraintsto constrain or optimise various degrees of freedom.
To minimise energy and forces we typically use an optimisation algorithm
- With a zero temperature thermostat we would eventually expect the structure to reach some local minimum energy in the potential-energy surface.
- Generally it is more efficient to achieve this by a dedicated “geometry optimisation” algorithm that seeks to minimise energy and forces.
- Let’s grab a molecular structure and apply a semi-empirical potential to calculate forces.
from ase.calculators.mopac import MOPAC
atoms = ase.build.molecule('CH3CH2OH')
atoms.calc = MOPAC()
atoms.get_forces()
MOPAC Job: "mopac.mop" ended normally on Apr 17, 2023, at 11:15.
array([[ 0.05314722, -0.14217078, 0. ],
[ 0.08516519, 0.2916233 , 0. ],
[ 0.46667555, 0.31685049, 0. ],
[-0.30313837, -0.04753196, 0. ],
[-0.29917533, 0.04623103, 0.23729349],
[-0.2992056 , 0.04519975, -0.23729349],
[ 0.11622196, -0.06459161, 0. ],
[ 0.09013888, -0.22333767, 0.08025091],
[ 0.09017054, -0.22227256, -0.08025091]])
There are a range of algorthms in the ase.optimize module
- We can see from the forces that this potential does not quite agree with the structure from the G2 database.
- For further calculations using MOPAC we need to find the local minimum.
- ASE provides a range of algorthms in the ase.optimize module.
- There are three preparation steps:
- Build the
Atomsstructure - Attach a
Calculatorobject - Instantiate the optimisation object with the
Atomsobject
- Build the
from ase.optimize import GPMin
# To avoid that "ended normally" message on every step, we
# tweak the command a bit
atoms = ase.build.molecule('CH3CH2OH')
atoms.calc = MOPAC(command='mopac PREFIX.mop 1> /dev/null')
opt = GPMin(atoms)
- When we run the optimisation the original
Atomsobject updates its positions.
opt.run(fmax=1e-3)
atoms.get_forces()
Step Time Energy fmax
GPMin: 0 11:15:28 -2.385274 0.564075
GPMin: 1 11:15:28 -2.392905 1.033572
GPMin: 2 11:15:28 -2.403956 0.742120
GPMin: 3 11:15:28 -2.420694 0.221260
GPMin: 4 11:15:28 -2.421733 0.151170
GPMin: 5 11:15:28 -2.422788 0.015994
GPMin: 6 11:15:28 -2.422797 0.010651
GPMin: 7 11:15:28 -2.422799 0.006327
GPMin: 8 11:15:28 -2.422801 0.003863
GPMin: 9 11:15:28 -2.422801 0.000485
array([[ 7.17242279e-05, 1.52121276e-04, -2.75015147e-04],
[ 2.59490798e-04, 9.01106079e-05, 4.03286166e-06],
[-4.31949839e-04, -1.41150158e-04, 1.70377564e-04],
[ 1.22850506e-04, -3.39324113e-04, 3.42142780e-05],
[-6.14902993e-05, 4.04153448e-05, -5.03023605e-05],
[-1.96786304e-04, -1.73499780e-04, -1.32824250e-04],
[ 1.40066056e-05, 1.05244680e-04, 2.27704910e-04],
[-3.03548727e-05, -6.07097455e-05, 8.32590795e-06],
[ 2.52552541e-04, 3.26878615e-04, 1.34862363e-05]])
Exercise: Comparing optimisers
Compare the results and performance of QuasiNewton, FIRE and GPMin optimizers for a few small organic molecules.
We can apply constraints to the Atoms object before optimisation
- We don’t always want to optimize every degree of freedom; perhaps we can assume some region is rigid, or we are using a forcefield that fixes some bond length.
- In ASE this is achieved by applying constraints to the Atoms object before optimisation.
- Suppose we want to explore the effect of stretching the C-C bond in ethane, while allowing the hydrogen atoms to relax to their local minimum at each configuration.
- The steps are largely the same as in the previous calculation
- Build the
Atomsstructure (build_stretched_ethane) - Apply a constraint (
build_stretched_ethane) - Attach a calculator (
get_optimised_energy) - Create an optimisation object (
get_optimised_energy) - Run the optimisation routine (
get_optimised_energy)
- Build the
- Python list comprehension is used to repeat the process for various C-C bond lengths
from ase.constraints import FixAtoms
import numpy as np
ethane = ase.build.molecule('C2H6')
g2_c_c = ethane.positions[1, 2] - ethane.positions[0, 2]
constraint = FixAtoms(mask=(atoms.symbols == 'C'))
def build_stretched_ethane(d: float = g2_c_c) -> Atoms:
shift = (d - g2_c_c) / 2
ethane = ase.build.molecule('C2H6')
ethane.positions[[0, 2, 3, 4]] += [0, 0, shift]
ethane.positions[[1, 5, 6, 7]] -= [0, 0, shift]
ethane.set_constraint(constraint)
return ethane
def get_optimised_energy(atoms: Atoms) -> float:
# Redirect STDOUT to avoid annoying message on every step
atoms.calc = MOPAC(command='mopac PREFIX.mop 1> /dev/null')
opt = GPMin(atoms)
opt.run(fmax=1e-3)
return atoms.get_potential_energy()
lengths = np.linspace(0.8, 1.04, 10) * g2_c_c
energies = [get_optimised_energy(build_stretched_ethane(d)) for d in lengths]
Step Time Energy fmax
GPMin: 0 11:15:29 0.208544 0.693187
GPMin: 1 11:15:29 0.010296 0.115406
GPMin: 2 11:15:29 0.008034 0.024697
GPMin: 3 11:15:29 0.007877 0.011408
GPMin: 4 11:15:29 0.007868 0.001131
GPMin: 5 11:15:29 0.007871 0.000784
Step Time Energy fmax
GPMin: 0 11:15:29 0.004891 0.621748
GPMin: 1 11:15:29 -0.148598 0.110453
GPMin: 2 11:15:29 -0.151673 0.029460
GPMin: 3 11:15:29 -0.151831 0.010914
GPMin: 4 11:15:29 -0.151840 0.001365
GPMin: 5 11:15:30 -0.151837 0.000968
Step Time Energy fmax
GPMin: 0 11:15:30 -0.185557 0.542410
GPMin: 1 11:15:30 -0.297151 0.110753
GPMin: 2 11:15:30 -0.301405 0.033660
GPMin: 3 11:15:30 -0.301558 0.011636
GPMin: 4 11:15:30 -0.301564 0.006232
GPMin: 5 11:15:30 -0.301566 0.001605
GPMin: 6 11:15:30 -0.301564 0.000377
Step Time Energy fmax
GPMin: 0 11:15:30 -0.359554 0.454383
GPMin: 1 11:15:30 -0.433351 0.115860
GPMin: 2 11:15:30 -0.438910 0.036632
GPMin: 3 11:15:30 -0.439056 0.011921
GPMin: 4 11:15:30 -0.439063 0.001158
GPMin: 5 11:15:30 -0.439061 0.000215
Step Time Energy fmax
GPMin: 0 11:15:30 -0.512800 0.356939
GPMin: 1 11:15:30 -0.554601 0.120804
GPMin: 2 11:15:30 -0.561024 0.037269
GPMin: 3 11:15:30 -0.561161 0.009839
GPMin: 4 11:15:30 -0.561165 0.001118
GPMin: 5 11:15:31 -0.561165 0.000591
Step Time Energy fmax
GPMin: 0 11:15:31 -0.639633 0.249621
GPMin: 1 11:15:31 -0.657675 0.114301
GPMin: 2 11:15:31 -0.663367 0.034829
GPMin: 3 11:15:31 -0.663479 0.006902
GPMin: 4 11:15:31 -0.663481 0.001379
GPMin: 5 11:15:31 -0.663481 0.000663
Step Time Energy fmax
GPMin: 0 11:15:31 -0.732713 0.133629
GPMin: 1 11:15:31 -0.738455 0.063419
GPMin: 2 11:15:31 -0.740055 0.018883
GPMin: 3 11:15:31 -0.740080 0.007060
GPMin: 4 11:15:31 -0.740082 0.000661
Step Time Energy fmax
GPMin: 0 11:15:31 -0.782718 0.048157
GPMin: 1 11:15:31 -0.783098 0.022657
GPMin: 2 11:15:31 -0.783185 0.003206
GPMin: 3 11:15:31 -0.783185 0.000318
Step Time Energy fmax
GPMin: 0 11:15:31 -0.778047 0.160836
GPMin: 1 11:15:31 -0.782210 0.063283
GPMin: 2 11:15:31 -0.782847 0.007287
GPMin: 3 11:15:31 -0.782853 0.000394
Step Time Energy fmax
GPMin: 0 11:15:31 -0.704544 0.316270
GPMin: 1 11:15:31 -0.706982 0.214952
GPMin: 2 11:15:31 -0.726086 0.073858
GPMin: 3 11:15:31 -0.726699 0.006312
GPMin: 4 11:15:31 -0.726703 0.001655
GPMin: 5 11:15:31 -0.726703 0.000322
%matplotlib inline
import matplotlib.pyplot as plt
plt.plot(lengths, energies, 'o-')
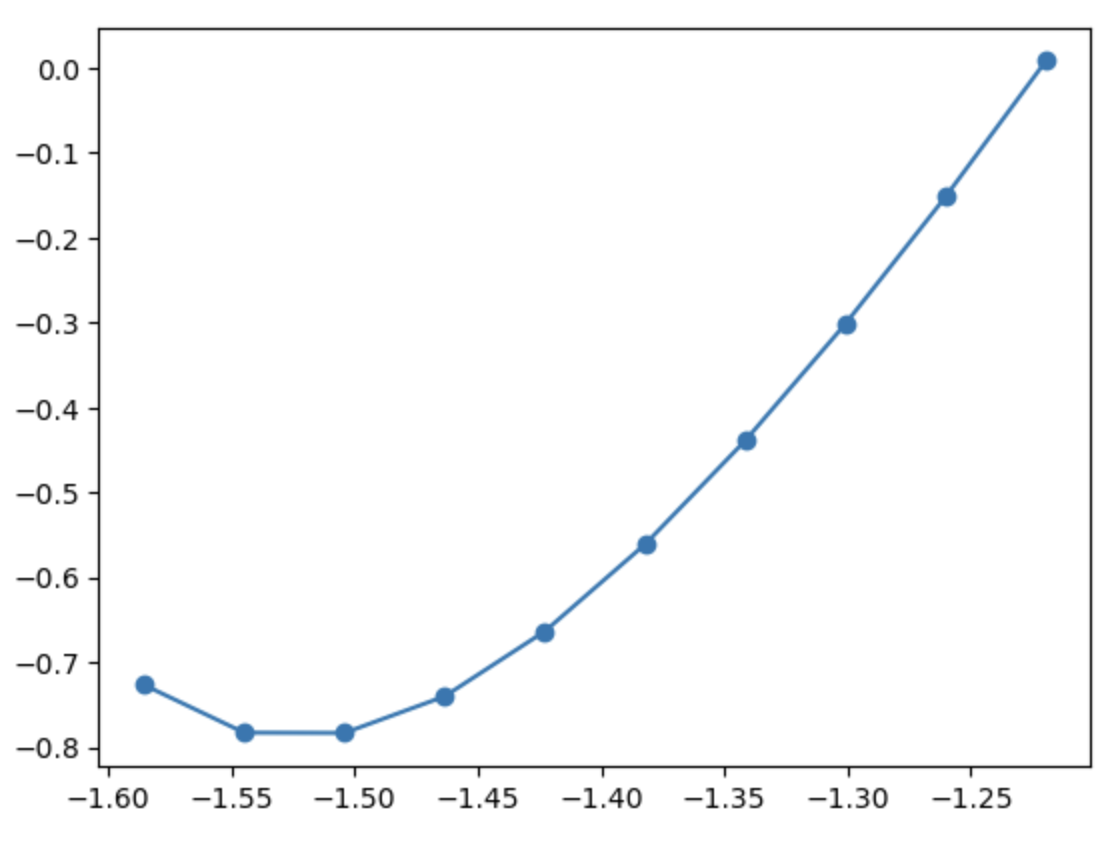
Filters can be used to optimise other degrees of freedom
- By default the geometry optimization only considers the atomic positions.
- To present other degrees of freedom, ASE includes “filters” that present them to the optimizer.
- Suppose we want to find the minimum-energy Si lattice parameters for a GAP machine-learned potential.
- First lets set up the
Atomsobject and attach the calculator
Note
More information on the Quippy
Potentialobject for GAP can be found in the External Calculators episode.
from ase.constraints import StrainFilter
from pathlib import Path
from quippy.potential import Potential
gap = Potential(param_filename=str(Path.cwd() / 'Si_PRX_GAP/gp_iter6_sparse9k.xml'))
si = ase.build.bulk('Si', cubic=True)
si.calc = gap
Filters place different information into the Atoms properties interface
- The filter presents a similar interface to Atoms, but “cheats” and puts different information into the positions data.
- In this case
positionis the a strain relative to the initial unit cell, and stresses are remapped toforces.
si_strain = StrainFilter(si)
print(si_strain.get_positions())
print(si_strain.get_forces())
array([[0., 0., 0.],
[0., 0., 0.]])
array([[ 1.55837119e+00, 1.55837119e+00, 1.55837119e+00],
[ 7.06032455e-16, -6.41847686e-17, -8.67361738e-18]])
- To get a Si lattice with very low stress we run the optimisation with the filter as the
Atomsobject:
from ase.optimize import QuasiNewton
opt = QuasiNewton(si_strain)
opt.run(fmax=1e-4)
print(f"Lattice parameter: {si.cell.lengths()[0]}")
print("Residual stress:")
print(si.get_stress())
Step[ FC] Time Energy fmax
*Force-consistent energies used in optimization.
BFGSLineSearch: 0[ 0] 11:15:35 -1305.407858* 2.6992
BFGSLineSearch: 1[ 2] 11:15:36 -1305.420818* 0.3445
BFGSLineSearch: 2[ 3] 11:15:36 -1305.421038* 0.0080
BFGSLineSearch: 3[ 4] 11:15:36 -1305.421038* 0.0000
Lattice parameter: 5.460914996056226
Residual stress:
[-8.62027513e-08 -8.62027472e-08 -8.62027630e-08 -2.58637937e-15
-6.90550987e-15 -4.46639070e-15]
- We can optimise the lattice vectors and positions at the same time using a UnitCellFilter or the more sophisticated ExpCellFilter.
MD and geometry optimization can be combined to find glassy local-minimum phases
- Let’s generate an amorphous Si cell by i) stretching a supercell and running a few steps of high-temperature MD; ii) performing a local optimisation to find a glassy local-minimum phase.
- Step i) is a similar process to the Cu calculation in the previous episode, but now we are keeping the periodic boundary and using a machine-learned potential.
- It should take a few minutes to run.
def printenergy(atoms: Atoms) -> None:
"""Function to print the potential, kinetic and total energy"""
epot = atoms.get_potential_energy() / len(atoms)
ekin = atoms.get_kinetic_energy() / len(atoms)
temperature = ekin / (1.5 * units.kB)
print(f'Energy per atom: Epot = {epot:.3f}eV Ekin = {ekin:.3f}eV '
f'(T={temperature:3.0f}K) Etot = {epot+ekin:.3f}eV')
def energy_observer():
printenergy(si)
si = ase.build.bulk('Si', cubic=True) * [2, 2, 2]
si.set_cell(si.cell * 1.2, scale_atoms=True)
si.calc = gap
si.calc.name = 'Si melt'
MaxwellBoltzmannDistribution(si, temperature_K=3000)
dyn = Langevin(si, 5 * ase.units.fs, friction=0.002, temperature_K=3000)
dyn.attach(energy_observer, interval=20)
traj = Trajectory('si_melt.traj', 'w', si)
dyn.attach(traj.write, interval=20)
dyn.run(400)
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
Energy per atom: Epot = 0.457eV Ekin = 0.055eV (T=423K) Etot = 0.512eV
- We now optimise lattice vectors and atom positions using the
ExpCellFilter.
Discussion
Note that the trajectory writer is directed to the original Si Atoms object, not the filtered version. Why?.
from ase.constraints import ExpCellFilter
si_expcell = ExpCellFilter(si)
opt = QuasiNewton(si_expcell)
traj = Trajectory('si_glass.traj', 'w', si)
opt.attach(traj)
opt.run(fmax=1e-3)
Step[ FC] Time Energy fmax
*Force-consistent energies used in optimization.
BFGSLineSearch: 0[ 0] 11:23:00 -10438.560479* 0.0082
BFGSLineSearch: 1[ 2] 11:23:02 -10438.560479* 0.0083
BFGSLineSearch: 2[ 4] 11:23:05 -10438.560479* 0.0021
BFGSLineSearch: 3[ 6] 11:23:07 -10438.560479* 0.0130
BFGSLineSearch: 4[ 8] 11:23:10 -10438.560479* 0.0008
- We can confirm that the forces and stresses are low, so we should be in a local minimum.
print("Max force:")
print(np.linalg.norm(si.get_forces(), axis=1).max())
print("Stress:")
print(si.get_stress())
Max force:
0.000764832698961055
Stress:
[ 1.17734167e-07 5.24076825e-08 -1.70158721e-07 -1.69164613e-08
-3.43769568e-08 -9.34363441e-09]
Exercise: Comparing crystal and amorphous Si
Compare the energy and lattice vectors of the amorphous and crystalline Si.
Exercise: Fixing the symmetry
A very useful constraint is the FixSymmetry class. Find a geometry file for a high-symmetry crystal structure. With a Calculator of your choice, perform a geometry optimization of the lattice vectors and atomic positions while fixing the symmetry. If compute power allows, you may choose to use DFT. If using Quantum Espresso, you may find it helpful to look at the SocketIOCalculator setup in the extra content section.
Key Points
To minimise energy and forces we typically use an optimisation algorithm
There are a range of algorthms in the
ase.optimizemoduleWe can apply constraints to the
Atomsobject before optimisationFilters can be used to optimise other degrees of freedom
Filters place different information into the
Atomsproperties interfaceMD and geometry optimization can be combined to find glassy local-minimum phases