The Atoms Object
Overview
Teaching: 15 min
Exercises: 5 minQuestions
How can I describe a molecule or crystal using the
Atomsclass?How can I access and adjust
Atomsinformation?What built-in help is available from ASE?
Objectives
Create and visualise an
AtomsobjectReturn simple structural information from an
AtomsobjectAdjust the default properties for an
AtomsobjectAccess built-in help using
?and tab-completion
Code connection
In this episode we explore the
Atomsclass, which contains methods for getting and setting simple properties for a molecule or material.
Molecules and materials are represented by the Atoms class
- We can define a molecule with lists of symbols and positions:
from ase import Atoms
d = 1.10
molecule = Atoms(['N', 'N'], positions=[(0., 0., 0.), (0., 0., d)])
- For convenience we can compress the list of symbols to a chemical formula:
molecule = Atoms('N2', positions=[(0., 0., 0.), (0., 0., d)])
ase.visualize.view and nglview can be used to visualise an Atoms object
- It can be useful to visualise our structure to make sure it is reasonable.
ase.visualize.viewcan be used is able to display trajectories and sequences of structures, a feature which we will use in later tutorials.- The
nglviewbackend integrates nicely with Jupyter notebooks, so that structures are displayed “inline” rather than in a pop-out window. - With
nglviewleft-click-and-drag is used for rotation.
from ase.visualize import view
view(molecule, viewer='ngl')
To describe crystals we use the cell and pbc keywords
- Many interesting systems are crystals, described by atomic positions in a periodic unit cell.
- There are two relevant keyword settings for an
Atomsobject: the unit cell itself (cell) and the periodic boundary conditions (pbc). - If the cell is specified with three values it is assumed to be cubic.
- In other cases we might use the full 3x3 matrix to describe off-diagonal terms, e.g.
cell=[[a, -a, 0], [a, a, 0], [0, 0, a]] - Once a
cellis specified we can use thescaled_positionskeyword to specify atomic positions relative to lattice vectors. - We set
pbc=Trueto indicate periodic boundary conditions in all directions. - PBCs can also be specified along each direction, e.g.
pbc=[True, True, False]for a “slab” calculation with exposed surfaces.
Shortcuts
We have defined a Python function
show()which will show enlarged atoms and a unit cell withnglview. This will save us from writing four lines of code every time we want to display a crystal.
a = 5.387
crystal = Atoms('Zn4S4',
scaled_positions=[[0., 0., 0.],
[0., 0.5, 0.5],
[0.5, 0., 0.5],
[0.5, 0.5, 0.],
[0.25, 0.75, 0.75],
[0.25, 0.25, 0.25],
[0.75, 0.75, 0.25],
[0.75, 0.25, 0.75]],
cell=[a, a, a],
pbc=True)
view(crystal, viewer='ngl')
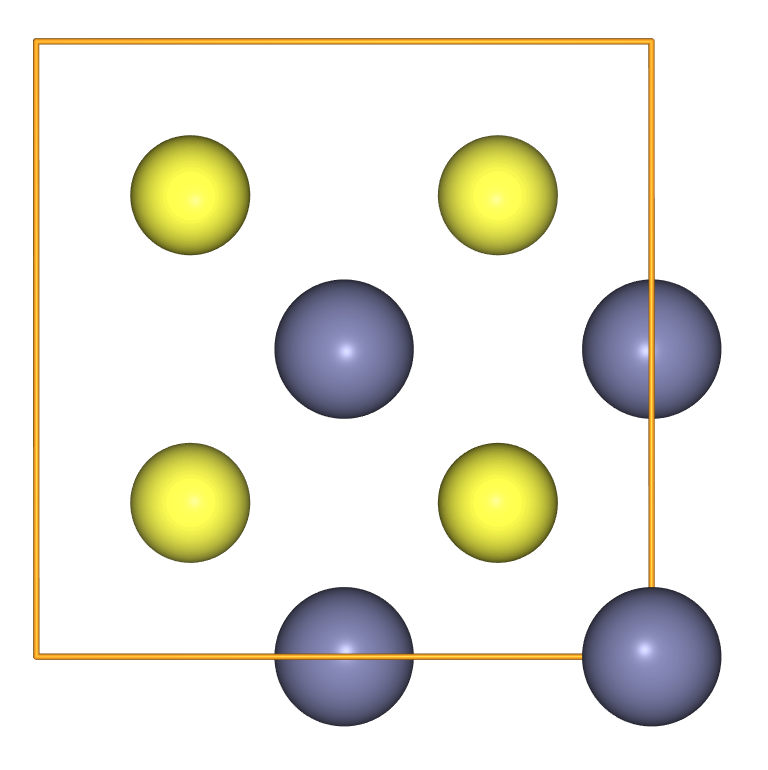
To access information from Atoms we use “getter” methods
- Now that we have some Atoms objects we can see what information is available from them.
- To access this information we call some “getter” methods on the
moleculeobject we created above. - Information about the chemical species and atomic positions is provided by us when we create the object.
print("N2 positions")
print(molecule.get_positions(), end="\n\n")
print("N2 symbols")
print(molecule.get_chemical_symbols(), end="\n\n")
N2 positions
[[0. 0. 0. ]
[0. 0. 1.1]]
N2 symbols
['N', 'N']
- There is also some information that is retrieved from a database, or derived from these database values.
print("N2 masses")
print(molecule.get_masses(), end="\n\n")
print("N2 center of mass")
print(molecule.get_center_of_mass())
N2 masses
[14.007 14.007]
N2 center of mass
[0. 0. 0.55]
We can override default keyword values when we create an Atoms object
- When we create an
Atomsobject we can override the defaultmassesand include some isotopic effects. - The center of mass will also be updated accordingly, as this is derived from
masses.
d = 1.10
isotope = Atoms('N2',
positions=[(0., 0., 0.), (0., 0., d)],
masses=[13.006, 14.003])
print("13N-14N masses")
print(isotope.get_masses(), end="\n\n")
print("13N-14N center of mass")
print(isotope.get_center_of_mass())
13N-14N masses
[13.006 14.003]
13N-14N center of mass
[0. 0. 0.57030249]
To adjust information in Atoms we use “setter” methods
- Once an
Atomsinstance is created we can override the defaultmassesusing “setter” methods. - In this example we start by copying the
moleculeobject with the default masses. - We reset the masses using the
set_masses()method, and the centre of mass is updated automatically.
isotope = molecule.copy()
print("Center of mass before modifying masses:")
print(isotope.get_center_of_mass(), end='\n\n')
isotope.set_masses([13.006, 14.003])
print("Center of mass after modifying masses:")
print(isotope.get_center_of_mass())
Center of mass before modifying masses:
[0. 0. 0.55]
Center of mass after modifying masses:
[0. 0. 0.57030249]
Accessing help
In a jupyter notebook or IPython terminal we can get the “docstring” of a method or function by adding ? to the name
isotope.get_center_of_mass?Signature: isotope.get_center_of_mass(scaled=False) Docstring: Get the center of mass. If scaled=True the center of mass in scaled coordinates is returned. File: ~/src/ase/ase/atoms.py Type: methodWe can also get access to the available methods and properties with tab-completion. In a Jupyter notebook or IPython terminal, try:
crystal.[TAB]where
[TAB]means “hit the TAB key”. You should see that theAtomsobject has a lot of features available; however not all of them will work until we start using theCalculatorclass.
Exercise: Atom features
Use tab-completion and docstrings to explore the features of
Atoms. In the ZnS structure, find the distance between the first Zn atom and the four S atoms. Are some of them nearer than others?Hint: you may need to use the “minimum image convention”
Key Points
Molecules and materials are represented by the
Atomsclass
ase.visualize.viewandnglviewcan be used to visualise anAtomsobjectTo describe crystals we use the
cellandpbckeywordsTo access information from
Atomswe use “getter” methodsWe can override default keyword values when we create an
AtomsobjectTo adjust information in
Atomswe use “setter” methods